 Première séquence vraiment complète d’un génome humain publiée par le consortium Telomere-to-Telomere (T2T), après des percées dans les nouvelles technologies (auparavant codées à un peu plus de 90 %)
Première séquence vraiment complète d’un génome humain publiée par le consortium Telomere-to-Telomere (T2T), après des percées dans les nouvelles technologies (auparavant codées à un peu plus de 90 %)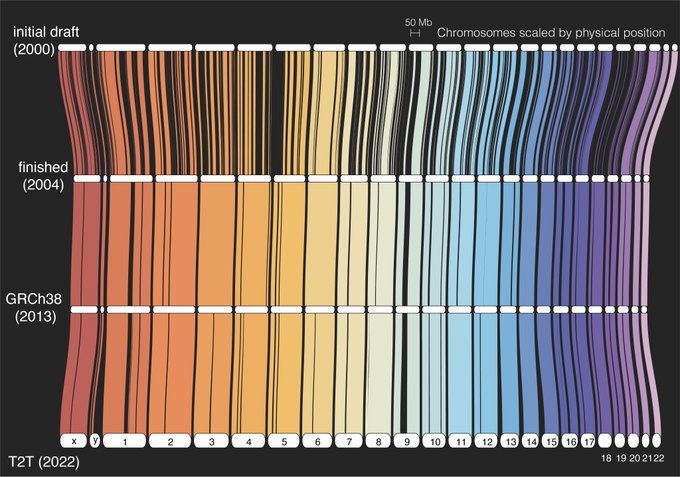
 La première séquence complète et sans interruption d’un génome humain.
La première séquence complète et sans interruption d’un génome humain. Infographie : Compléter la séquence du génome humain
Infographie : Compléter la séquence du génome humain Compléter la séquence du génome humain (encore)
Compléter la séquence du génome humain (encore)
Le consortium Telomere-to-Telomer vient de séquencer les 10 % délicats du génome humain essentiellement complet Une caractéristique fondamentale du domaine de la génomique est d’aspirer à l’exhaustivité. Après tout, la génomique est l’étude de tout l’ADN d’un organisme : son génome.
Une caractéristique fondamentale du domaine de la génomique est d’aspirer à l’exhaustivité. Après tout, la génomique est l’étude de tout l’ADN d’un organisme : son génome.
Les scientifiques du consortium Telomere-to-Telomere (T2T) ont maintenant rapporté la première séquence vraiment complète d’un génome humain, près de deux décennies après que le projet du génome humain a produit la première séquence (essentiellement complète) du génome humain. Les gens peuvent se demander comment il se fait que les scientifiques prétendent compléter à nouveau la séquence du génome humain . Ce n’était pas déjà fait ? Eh bien, oui et non.![]() Comprendre cette nouvelle étape nécessite une appréciation du produit de signature très nuancé et souvent mal compris de l’effort initial. Elle renvoie aussi à une ambition scientifique toujours recherchée mais rarement atteinte : le rêve d’achèvement.
Comprendre cette nouvelle étape nécessite une appréciation du produit de signature très nuancé et souvent mal compris de l’effort initial. Elle renvoie aussi à une ambition scientifique toujours recherchée mais rarement atteinte : le rêve d’achèvement.
تطبيقات #الجينوم_البشري طبية و علمية و اقتصادية و تقنية 🧬#Genomics #human_genome#Bioinformatics #T2T pic.twitter.com/OGCWVzW8J8
— د. عبدالمجيد الرفاعيAbdulmajeed Alrefaei (@DrAbdulmajeedFR) April 8, 2022
L’objectif principal du projet du génome humain était de générer la première séquence du génome humain. Alors que l’objectif était d’être aussi complet que possible, les chefs de projet ont mis des limites pratiques autour de cette philosophie ambitieuse et, en fait, n’envisageaient pas de poursuivre le projet jusqu’à ce que l’ordre des quelque trois milliards de bases d’ADN humain ait été séquencé. Cette approche raisonnée empêchait « la perfection de devenir l’ennemie du bien ».En 2001, le projet du génome humain et Celera Genomics ont chacun signalé la génération d’une ébauche de séquence du génome humain en grande pompe. Deux ans plus tard, en utilisant les meilleures technologies disponibles pour séquencer l’ADN et en les poussant à leurs limites absolues, le projet du génome humain a livré une séquence du génome humain d’ une qualité remarquable qui était presque complète , représentant plus de 90 % du génome humain. Il n’y avait pas de perspectives immédiates pour remplir les bits restants car les technologies de séquençage de l’ADN à l’époque n’étaient pas à la hauteur de la tâche.

 Mais certes, au milieu des célébrations exubérantes, quelques-uns de ces dirigeants ont peut-être semblé proclamer : « Nous avons terminé ; nous l’avons terminé; notre travail est terminé. Ces hyperboles étaient vraiment le champagne qui parlait. Il y avait des grognements à l’époque que de telles affirmations étaient malhonnêtes, mais je soutiens que les critiques voyaient le verre à moitié vide. Je considère la génération du projet de la première séquence du génome humain comme représentant un verre rempli à plus de 90 % ! Et ceux qui ont célébré bruyamment avaient mérité leur conversation imprécise sur le champagne.
Mais certes, au milieu des célébrations exubérantes, quelques-uns de ces dirigeants ont peut-être semblé proclamer : « Nous avons terminé ; nous l’avons terminé; notre travail est terminé. Ces hyperboles étaient vraiment le champagne qui parlait. Il y avait des grognements à l’époque que de telles affirmations étaient malhonnêtes, mais je soutiens que les critiques voyaient le verre à moitié vide. Je considère la génération du projet de la première séquence du génome humain comme représentant un verre rempli à plus de 90 % ! Et ceux qui ont célébré bruyamment avaient mérité leur conversation imprécise sur le champagne. La communauté de la génomique n’a jamais eu l’intention de simplement renoncer à terminer le travail. En fait, le développement de technologies et de stratégies de séquençage des régions difficiles restantes de l’ADN humain est rapidement devenu un domaine hautement prioritaire de la recherche en génomique . Ces efforts ont abouti à une suite de nouvelles technologies qui ont réduit le coût du séquençage de l’ADN de plus d’un million de fois.
La communauté de la génomique n’a jamais eu l’intention de simplement renoncer à terminer le travail. En fait, le développement de technologies et de stratégies de séquençage des régions difficiles restantes de l’ADN humain est rapidement devenu un domaine hautement prioritaire de la recherche en génomique . Ces efforts ont abouti à une suite de nouvelles technologies qui ont réduit le coût du séquençage de l’ADN de plus d’un million de fois.
Mais même avec ces nouvelles technologies, produire une séquence du génome humain de bout en bout est resté un problème très difficile, épuisant certains des chercheurs en génomique les plus talentueux
 La séquence nouvellement ajoutée, représentant près de 10% du génome humain, comprend certains gènes et de grandes quantités d’ADN répétitif, les régions génomiques les plus difficiles à séquencer. La majeure partie de cet ADN réside près des télomères répétitifs (les extrémités longues et arrière de chaque chromosome) et des centromères (la section médiane dense de chaque chromosome). Ces découvertes s’ajoutent à la connaissance croissante du génome humain, y compris des cartes plus précises pour cinq bras chromosomiques, et stimuleront de nouvelles lignes de recherche.
La séquence nouvellement ajoutée, représentant près de 10% du génome humain, comprend certains gènes et de grandes quantités d’ADN répétitif, les régions génomiques les plus difficiles à séquencer. La majeure partie de cet ADN réside près des télomères répétitifs (les extrémités longues et arrière de chaque chromosome) et des centromères (la section médiane dense de chaque chromosome). Ces découvertes s’ajoutent à la connaissance croissante du génome humain, y compris des cartes plus précises pour cinq bras chromosomiques, et stimuleront de nouvelles lignes de recherche.
Il est gratifiant de reconnaître comment les chercheurs du consortium T2T se sont appuyés sur les succès des génomiques du projet du génome humain qui les ont précédés et, plus tard, de ceux qui ont apporté de puissantes nouvelles technologies de séquençage de l’ADN et des stratégies de calcul innovantes pour relever le défi. Leurs efforts ont abouti à une séquence du génome humain vraiment complète, du télomère au télomère de chaque chromosome humain. Cette avancée nous rapproche toujours plus de l’avenir promis par la génomique. Pour la première fois, il semble possible que les chercheurs et les cliniciens soient éventuellement en mesure d’identifier toutes les variantes de l’ADN d’une personne et d’utiliser ces informations de manière globale pour mieux guider leurs soins de santé.
Cette avancée nous rapproche toujours plus de l’avenir promis par la génomique. Pour la première fois, il semble possible que les chercheurs et les cliniciens soient éventuellement en mesure d’identifier toutes les variantes de l’ADN d’une personne et d’utiliser ces informations de manière globale pour mieux guider leurs soins de santé.
J’applaudis énergiquement aux réalisations du consortium T2T. L’humanité vous sera éternellement reconnaissante pour votre persévérance, votre créativité, votre énergie et votre esprit d’aventure. Votre cadeau est la première représentation complète du schéma directeur de l’ADN humain, qui catalysera les avancées futures de la génomique, de la biologie humaine et de la médecine . En même temps, et avec le même esprit ambitieux d’être complet en génomique, permettez-moi d’admettre volontiers que notre travail n’est toujours pas terminé. Avec une séquence complète du génome humain maintenant en main, notre travail se poursuit pour comprendre comment le génome humain fonctionne, comment nos génomes diffèrent les uns des autres (y compris à travers les nombreuses populations diverses du monde), comment ces différences influencent notre santé et comment les informations sur ces différences peuvent être utilisées pour améliorer la pratique de la médecine.
En même temps, et avec le même esprit ambitieux d’être complet en génomique, permettez-moi d’admettre volontiers que notre travail n’est toujours pas terminé. Avec une séquence complète du génome humain maintenant en main, notre travail se poursuit pour comprendre comment le génome humain fonctionne, comment nos génomes diffèrent les uns des autres (y compris à travers les nombreuses populations diverses du monde), comment ces différences influencent notre santé et comment les informations sur ces différences peuvent être utilisées pour améliorer la pratique de la médecine.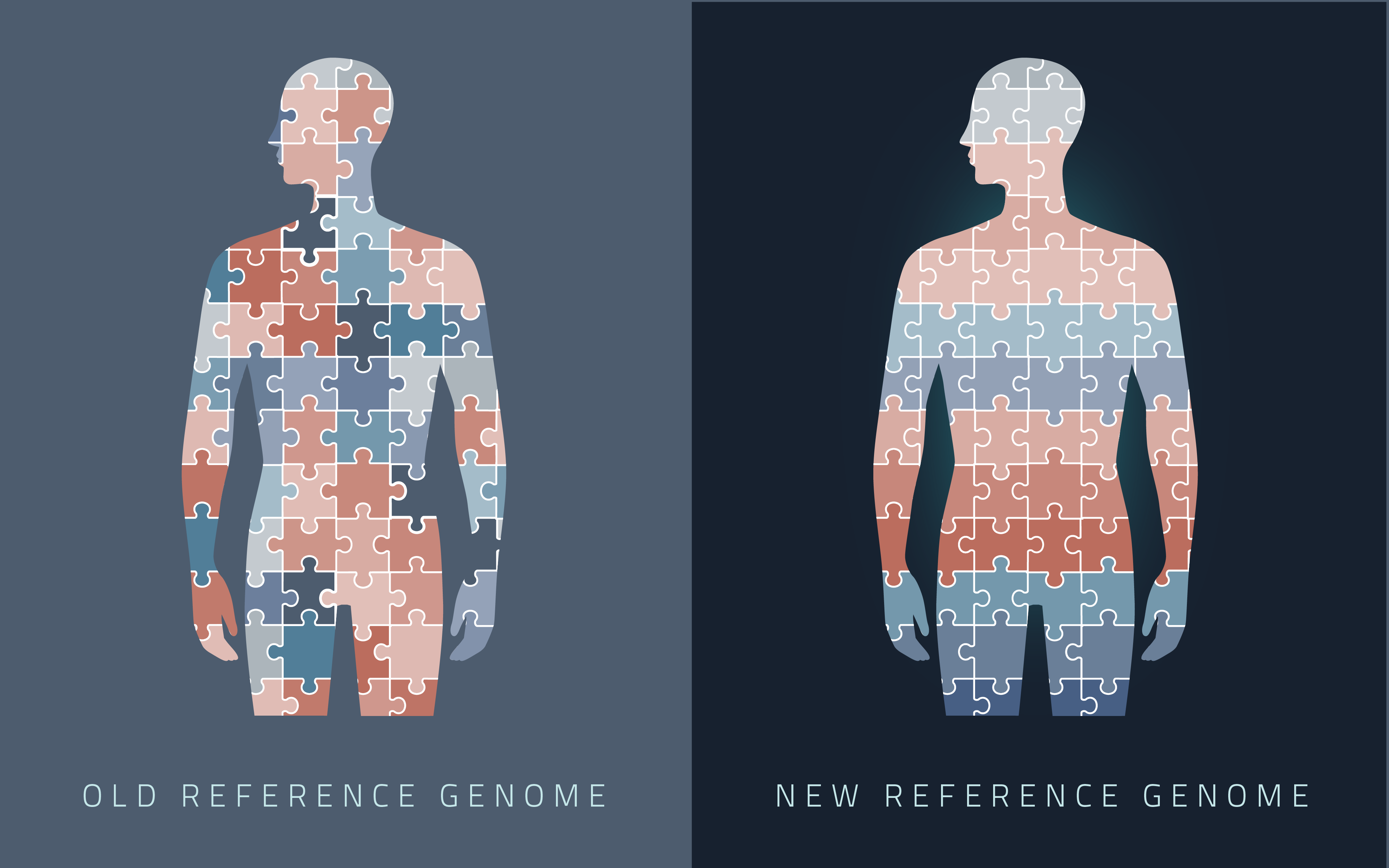 C’est la joie de la science et de la recherche : le travail n’est jamais terminé. Chaque avancée ouvre de nouvelles perspectives d’opportunités avec le sentiment fréquent que le meilleur reste à venir. Je continue certainement de croire que c’est vrai pour la génomique humaine, et je suis impatient de voir ce que la prochaine technologie, le prochain chercheur et le prochain consortium apporteront. Je me trouve à la fois fier et satisfait de ce que la génomique nous a apporté à ce jour : une combinaison d’essentiellement complet, vraiment complet et autrement.
C’est la joie de la science et de la recherche : le travail n’est jamais terminé. Chaque avancée ouvre de nouvelles perspectives d’opportunités avec le sentiment fréquent que le meilleur reste à venir. Je continue certainement de croire que c’est vrai pour la génomique humaine, et je suis impatient de voir ce que la prochaine technologie, le prochain chercheur et le prochain consortium apporteront. Je me trouve à la fois fier et satisfait de ce que la génomique nous a apporté à ce jour : une combinaison d’essentiellement complet, vraiment complet et autrement.
 Depuis sa publication initiale en 2000, le génome humain de référence n’a couvert que la fraction euchromatique du génome, laissant d’importantes régions hétérochromatiques inachevées. S’adressant aux 8 % restants du génome, le Consortium Telomere-to-Telomere (T2T) présente une séquence complète de 3,055 milliards de paires de bases d’un génome humain, T2T-CHM13, qui comprend des assemblages sans interruption pour tous les chromosomes sauf Y, corrige les erreurs dans les références antérieures, et introduit près de 200 millions de paires de bases de séquence contenant 1956 prédictions de gènes, dont 99 sont prédites comme codant pour des protéines. Les régions complétées comprennent tous les réseaux satellites centromériques, les duplications segmentaires récentes et les bras courts des cinq chromosomes acrocentriques, ouvrant ces régions complexes du génome à des études variationnelles et fonctionnelles.
Depuis sa publication initiale en 2000, le génome humain de référence n’a couvert que la fraction euchromatique du génome, laissant d’importantes régions hétérochromatiques inachevées. S’adressant aux 8 % restants du génome, le Consortium Telomere-to-Telomere (T2T) présente une séquence complète de 3,055 milliards de paires de bases d’un génome humain, T2T-CHM13, qui comprend des assemblages sans interruption pour tous les chromosomes sauf Y, corrige les erreurs dans les références antérieures, et introduit près de 200 millions de paires de bases de séquence contenant 1956 prédictions de gènes, dont 99 sont prédites comme codant pour des protéines. Les régions complétées comprennent tous les réseaux satellites centromériques, les duplications segmentaires récentes et les bras courts des cinq chromosomes acrocentriques, ouvrant ces régions complexes du génome à des études variationnelles et fonctionnelles. La première séquence complète et sans interruption d’un génome humain révèle des régions cachées
La première séquence complète et sans interruption d’un génome humain révèle des régions cachées
Des parties du génome humain désormais disponibles pour être étudiées pour la première fois sont importantes pour comprendre les maladies génétiques, la diversité humaine et l’évolution. La première séquence vraiment complète d’un génome humain, couvrant chaque chromosome de bout en bout sans lacunes et avec une précision sans précédent, est désormais accessible via le navigateur de génomes UCSC et est décrite dans six articles publiés le 31 mars dans Science .
La première séquence vraiment complète d’un génome humain, couvrant chaque chromosome de bout en bout sans lacunes et avec une précision sans précédent, est désormais accessible via le navigateur de génomes UCSC et est décrite dans six articles publiés le 31 mars dans Science . Depuis que le premier projet de travail d’une séquence du génome humain a été assemblé à l’UC Santa Cruz en 2000, la recherche en génomique a conduit à d’énormes progrès dans notre compréhension de la biologie et des maladies humaines. Néanmoins, des régions cruciales représentant environ 8 % du génome humain sont restées cachées aux scientifiques pendant plus de 20 ans en raison des limites des technologies de séquençage de l’ADN.
Depuis que le premier projet de travail d’une séquence du génome humain a été assemblé à l’UC Santa Cruz en 2000, la recherche en génomique a conduit à d’énormes progrès dans notre compréhension de la biologie et des maladies humaines. Néanmoins, des régions cruciales représentant environ 8 % du génome humain sont restées cachées aux scientifiques pendant plus de 20 ans en raison des limites des technologies de séquençage de l’ADN.
En 2019, Karen Miga, professeure adjointe d’ingénierie biomoléculaire à l’UC Santa Cruz, et Adam Phillippy de l’Institut national de recherche sur le génome humain (NHGRI) ont organisé une équipe internationale de scientifiques – le consortium Telomere-to-Telomere (T2T) – pour remplir les pièces manquantes. Leurs efforts ont maintenant porté leurs fruits. Le nouveau génome de référence, appelé T2T-CHM13, ajoute près de 200 millions de paires de bases de nouvelles séquences d’ADN, dont 99 gènes susceptibles de coder pour des protéines et près de 2 000 gènes candidats qui nécessitent une étude plus approfondie. Il corrige également des milliers d’erreurs structurelles dans la séquence de référence actuelle.
Le nouveau génome de référence, appelé T2T-CHM13, ajoute près de 200 millions de paires de bases de nouvelles séquences d’ADN, dont 99 gènes susceptibles de coder pour des protéines et près de 2 000 gènes candidats qui nécessitent une étude plus approfondie. Il corrige également des milliers d’erreurs structurelles dans la séquence de référence actuelle.
Les lacunes désormais comblées par la nouvelle séquence comprennent les bras courts entiers de cinq chromosomes humains et couvrent certaines des régions les plus complexes du génome. Celles-ci incluent des séquences d’ADN hautement répétitives trouvées dans et autour d’importantes structures chromosomiques telles que les télomères aux extrémités des chromosomes et les centromères qui coordonnent la séparation des chromosomes répliqués lors de la division cellulaire. La nouvelle séquence révèle également des duplications segmentaires non détectées auparavant, de longues étendues d’ADN qui sont dupliquées dans le génome et sont connues pour jouer un rôle important dans l’évolution et la maladie. « Ces parties du génome humain que nous n’avons pas pu étudier depuis plus de 20 ans sont importantes pour notre compréhension du fonctionnement du génome, des maladies génétiques, de la diversité et de l’évolution humaines », a déclaré Miga.
« Ces parties du génome humain que nous n’avons pas pu étudier depuis plus de 20 ans sont importantes pour notre compréhension du fonctionnement du génome, des maladies génétiques, de la diversité et de l’évolution humaines », a déclaré Miga.
De nombreuses régions nouvellement révélées ont des fonctions importantes dans le génome même si elles ne comprennent pas de gènes actifs.
Sommet de la montagne

« Nous voulions diffuser les informations d’une manière accessible et familière aux chercheurs afin qu’ils puissent commencer à s’en servir et utiliser tous les outils et ressources fournis par le navigateur », a expliqué Miga.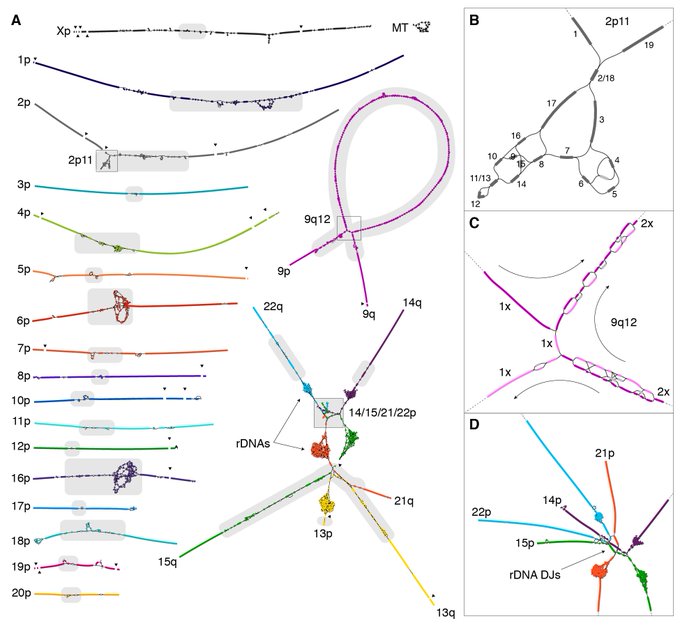
« Nous ajoutons un deuxième génome complet, et il y en aura d’autres », a expliqué Haussler. « La phase suivante consiste à penser que la référence du génome de l’humanité n’est pas une séquence unique du génome. C’est une transition profonde, le signe avant-coureur d’une nouvelle ère dans laquelle nous finirons par capturer la diversité humaine de manière impartiale.
Référence du pangénome humain Le Consortium T2T s’est désormais associé au Human Pangenome Reference Consortium , qui vise à créer une nouvelle « référence de pangénome humain » basée sur les séquences complètes du génome de 350 individus.
Le Consortium T2T s’est désormais associé au Human Pangenome Reference Consortium , qui vise à créer une nouvelle « référence de pangénome humain » basée sur les séquences complètes du génome de 350 individus.
« La pangénomique consiste à capturer la diversité de la population humaine, et il s’agit également de s’assurer que nous avons correctement capturé l’ensemble du génome », a déclaré Benedict Paten, professeur agrégé d’ingénierie biomoléculaire à la Baskin School of Engineering de l’UCSC, co-auteur des articles T2T, et un chef de file de l’effort de pangénomique. « Sans disposer d’une carte de ces régions du génome difficiles à séquencer sur plusieurs individus, nous manquons une grande partie de la variation présente dans notre population. T2T nous permet d’examiner des centaines de génomes, d’un télomère à l’autre. Ça va être génial! »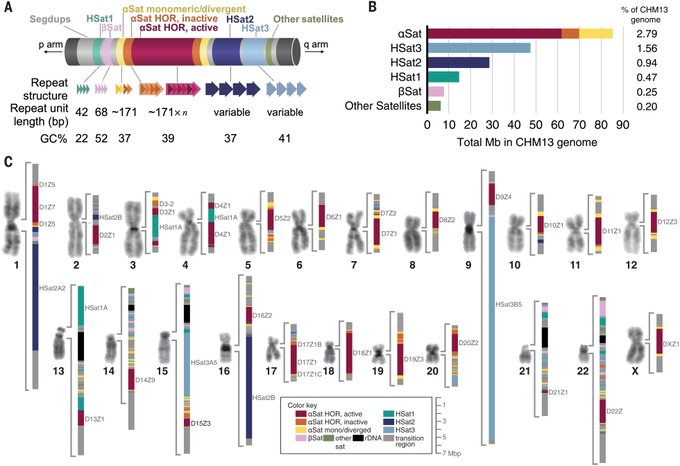
Variantes génétiques
La recherche de Miga s’est concentrée sur l’ADN satellite, les longues étendues de séquences d’ADN répétitives trouvées principalement dans et autour des télomères et des centromères. Les centromères séparent chaque chromosome en un bras court et un bras long et maintiennent ensemble les chromosomes dupliqués avant la division cellulaire.
« Les centromères jouent un rôle essentiel dans la manière dont les chromosomes se séparent correctement lors de la division cellulaire, et nous savons depuis un certain temps maintenant qu’ils sont mal régulés dans toutes sortes de maladies humaines. Mais nous n’avons jamais été en mesure de les étudier au niveau de la séquence. « , a déclaré Miga. « De loin, la plus grande partie des nouvelles séquences ajoutées à la référence sont des ADN satellites centromères. Pour la première fois, nous pouvons étudier « base par base » les séquences qui définissent le centromère et commencer à comprendre son fonctionnement. » Les technologies de séquençage de l’ADN « à lecture longue », telles que le séquençage des nanopores mis au point à l’UC Santa Cruz , étaient des outils essentiels pour le consortium T2T. Deux ensembles de données de séquençage à lecture longue – lectures haute fidélité (données HiFi des systèmes PacBio) et lectures extrêmement longues qui atteignent régulièrement des longueurs supérieures à 100 000 paires de bases (données ultra-longues des appareils Oxford Nanopore) – ont permis aux chercheurs de T2T de couvrir des régions répétitives et de développer stratégies pour s’assurer que l’assemblage était très précis. Miten Jain et d’autres chercheurs de l’UCSC Genomics Institute ont aidé à établir le protocole de lecture ultra-longue .
Les technologies de séquençage de l’ADN « à lecture longue », telles que le séquençage des nanopores mis au point à l’UC Santa Cruz , étaient des outils essentiels pour le consortium T2T. Deux ensembles de données de séquençage à lecture longue – lectures haute fidélité (données HiFi des systèmes PacBio) et lectures extrêmement longues qui atteignent régulièrement des longueurs supérieures à 100 000 paires de bases (données ultra-longues des appareils Oxford Nanopore) – ont permis aux chercheurs de T2T de couvrir des régions répétitives et de développer stratégies pour s’assurer que l’assemblage était très précis. Miten Jain et d’autres chercheurs de l’UCSC Genomics Institute ont aidé à établir le protocole de lecture ultra-longue .
Leaders en génomique
« Le travail de T2T reflète les efforts soutenus et dévoués de nombreuses personnes à l’UC Santa Cruz et ailleurs. Karen Miga a travaillé dur pour obtenir de véritables séquences de centromères dans les assemblages du génome humain pendant une décennie, et cela a finalement porté ses fruits ! » dit Kent. « Je suis très enthousiaste à l’idée de voir ce travail combiné aux efforts visant à obtenir des séquences de télomère à télomère d’autres ancêtres humains. Nous nous dirigeons rapidement vers une représentation vraiment complète du génome humain. Miga est co-auteur correspondant du principal article scientifique , » La séquence complète d’un génome humain « , avec Adam Phillippy au NHGRI et Evan Eichler à l’Université de Washington. Elle est également co-auteur correspondant des articles sur « Complete genomic and epigenetic maps of human centromeres » et « Epigenetic patterns in a complete human genome », et co-auteur des articles sur « Segmental duplications and their variation in a complete human genome ». génome », « Un génome de référence complet améliore l’analyse de la variation génétique humaine » et « Du télomère au télomère : l’état transcriptionnel et épigénétique des éléments répétés humains ».
Miga est co-auteur correspondant du principal article scientifique , » La séquence complète d’un génome humain « , avec Adam Phillippy au NHGRI et Evan Eichler à l’Université de Washington. Elle est également co-auteur correspondant des articles sur « Complete genomic and epigenetic maps of human centromeres » et « Epigenetic patterns in a complete human genome », et co-auteur des articles sur « Segmental duplications and their variation in a complete human genome ». génome », « Un génome de référence complet améliore l’analyse de la variation génétique humaine » et « Du télomère au télomère : l’état transcriptionnel et épigénétique des éléments répétés humains ».
Parmi les autres chercheurs de l’UC Santa Cruz Genomics Institute qui sont co-auteurs des articles figurent Benedict Paten, Mark Diekhans, Erik Garrison (maintenant au University of Tennessee Health Science Center), Marina Haukness, Miten Jain et Kishwar Shafin. Ce travail a été soutenu par les National Institutes of Health.
Télomère à télomère (T2T) La première séquence complète et sans interruption d’un génome humain.
La première séquence complète et sans interruption d’un génome humain.
Infographie : Compléter la séquence du génome humain Les chercheurs ont terminé une quête qui a commencé il y a 32 ans, après avoir découvert les dernières séquences d’ADN qui composent un génome humain. En savoir plus sur cette réalisation extraordinaire.
Les chercheurs ont terminé une quête qui a commencé il y a 32 ans, après avoir découvert les dernières séquences d’ADN qui composent un génome humain. En savoir plus sur cette réalisation extraordinaire.
Le projet du génome humain a pris fin en 2003, mais les chercheurs en génomique n’avaient pas encore déterminé chaque dernière base (ou lettre) de la séquence du génome humain. Au lieu de cela, ils n’avaient terminé qu’environ 92% de la séquence à ce moment-là. Pourquoi se sont-ils arrêtés là ?
Pourquoi était-il si difficile de terminer complètement la séquence du génome humain ?
Raison 1 : Le génome humain contient une quantité massive d’ADN.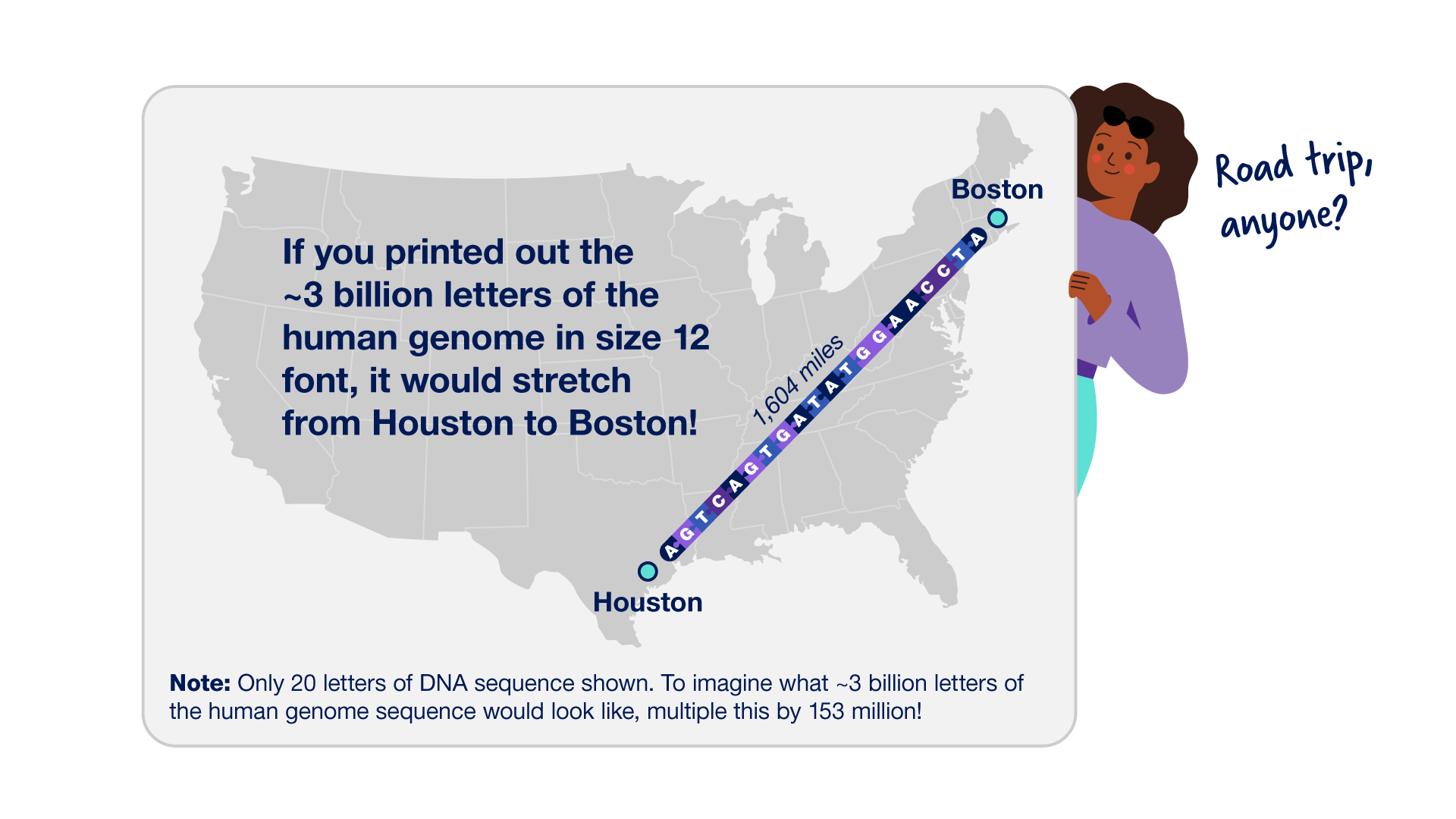 Le génome humain est constitué d’environ 3 milliards de bases dans un ordre précis, dont chacune peut être représentée par une lettre (G, A, T ou C). La séquence d’un génome ne peut pas être lue de bout en bout. Au lieu de cela, les chercheurs doivent d’abord déterminer la séquence de morceaux d’ADN aléatoires, puis utiliser ces séquences plus petites pour reconstituer l’ensemble de la séquence du génome comme un énorme puzzle.
Le génome humain est constitué d’environ 3 milliards de bases dans un ordre précis, dont chacune peut être représentée par une lettre (G, A, T ou C). La séquence d’un génome ne peut pas être lue de bout en bout. Au lieu de cela, les chercheurs doivent d’abord déterminer la séquence de morceaux d’ADN aléatoires, puis utiliser ces séquences plus petites pour reconstituer l’ensemble de la séquence du génome comme un énorme puzzle.
Raison 2 : Certaines parties de notre ADN sont douloureusement répétitives.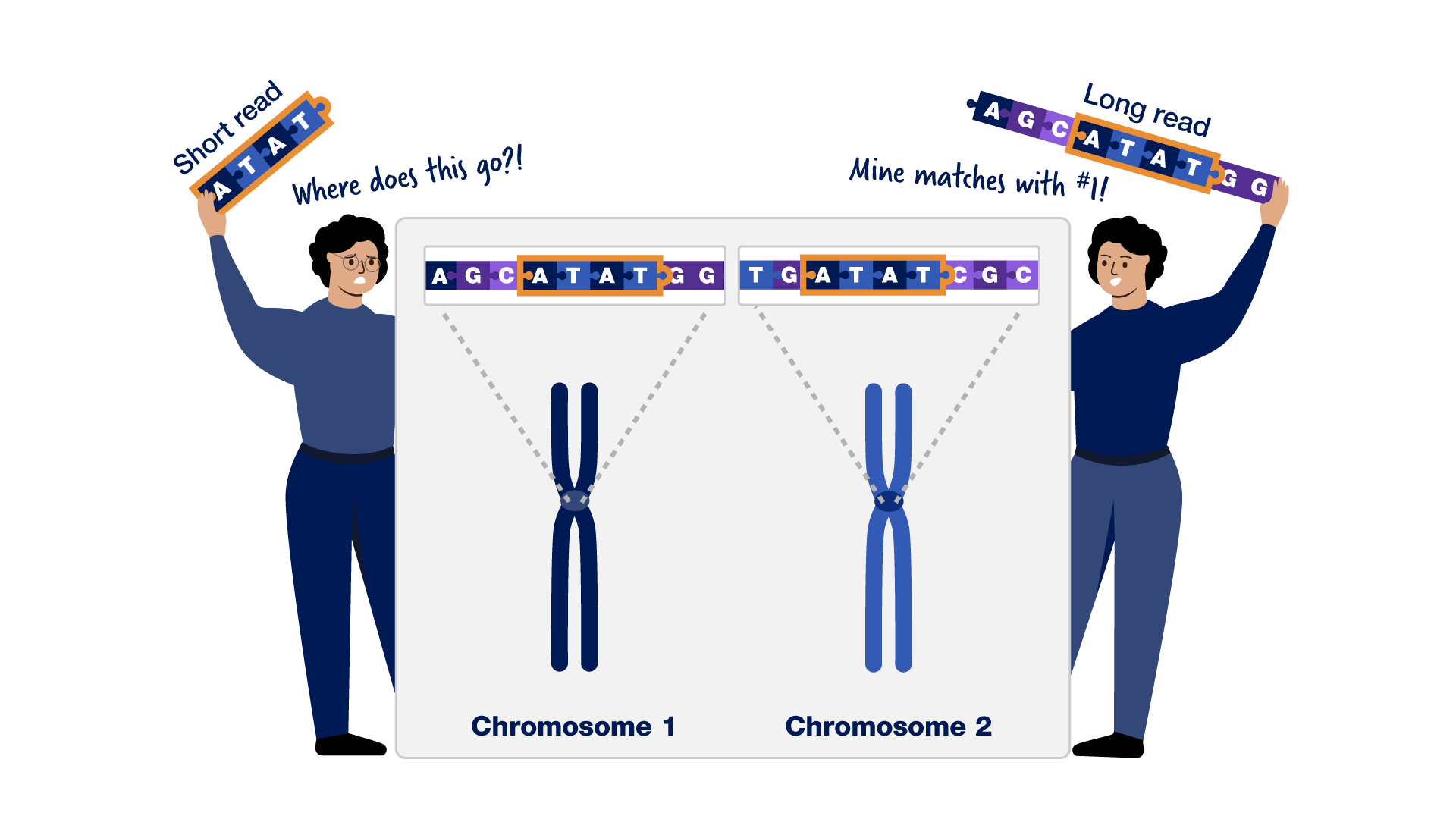 Certaines sections de la séquence du génome humain consistent en de longues séquences de lettres répétitives difficiles à placer au bon endroit. Au cours des deux dernières décennies, les chercheurs ont développé de nouvelles technologies pour lire de plus longues étendues d’ADN – de seulement environ 500 à maintenant plus de 100 000 lettres à la fois – ce qui leur a permis d’assembler toute la longueur des répétitions les plus difficiles.
Certaines sections de la séquence du génome humain consistent en de longues séquences de lettres répétitives difficiles à placer au bon endroit. Au cours des deux dernières décennies, les chercheurs ont développé de nouvelles technologies pour lire de plus longues étendues d’ADN – de seulement environ 500 à maintenant plus de 100 000 lettres à la fois – ce qui leur a permis d’assembler toute la longueur des répétitions les plus difficiles.
Raison 3 : Les premiers 92 % étaient difficiles. Les 8% restants étaient atroces.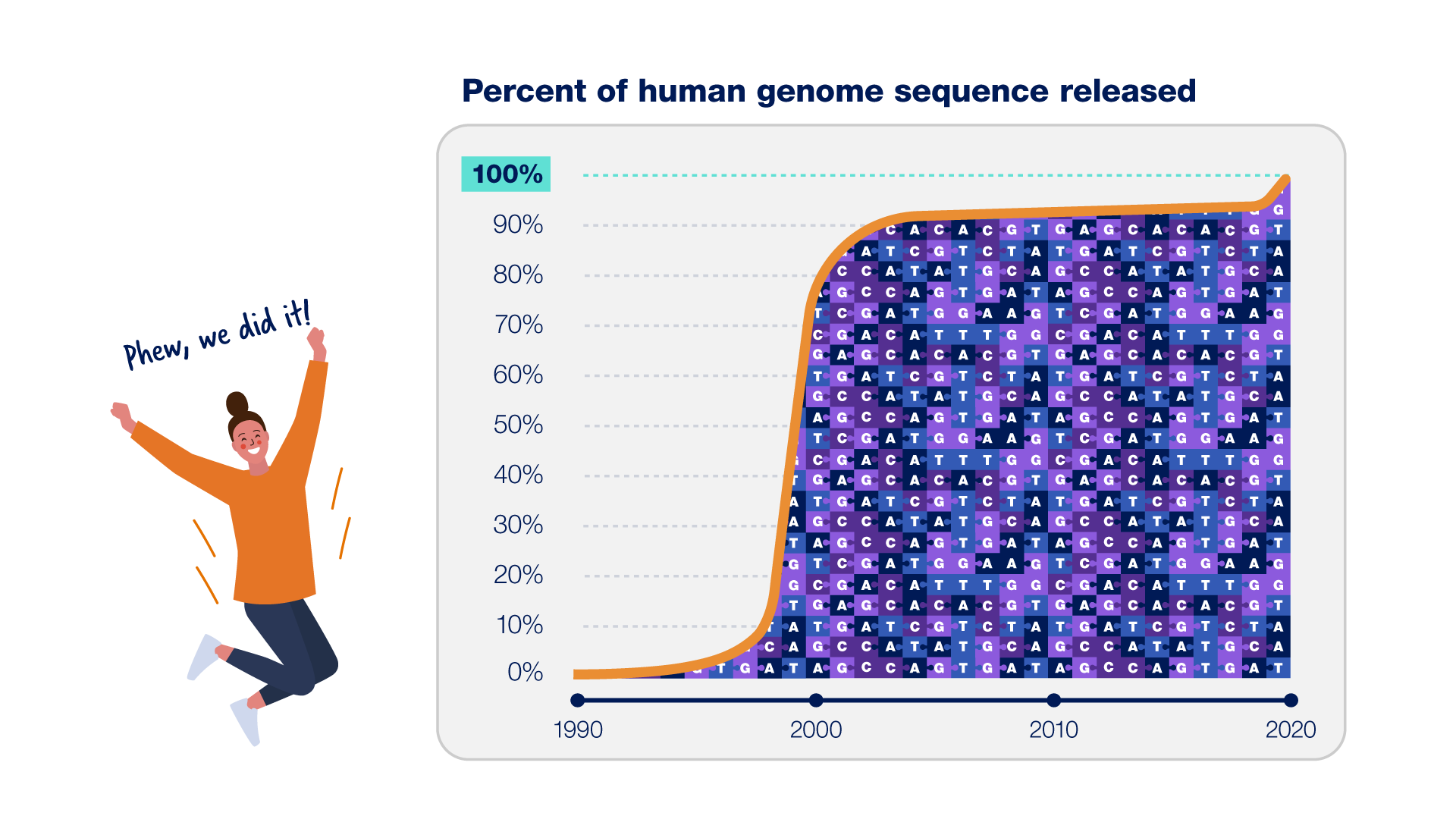 Ces répétitions d’ADN et d’autres obstacles se dressaient entre les chercheurs en génomique et les 8% finaux de la séquence du génome humain jusqu’à ce que de nouvelles technologies de laboratoire et de calcul soient développées. Il a fallu presque deux fois plus de temps pour terminer les derniers 8 % du génome humain que pour les premiers 92 % !
Ces répétitions d’ADN et d’autres obstacles se dressaient entre les chercheurs en génomique et les 8% finaux de la séquence du génome humain jusqu’à ce que de nouvelles technologies de laboratoire et de calcul soient développées. Il a fallu presque deux fois plus de temps pour terminer les derniers 8 % du génome humain que pour les premiers 92 % !
Raison 4 : Les 8 % restants avaient besoin d’une génération de chercheurs en génomique dévoués et dotés d’une vision.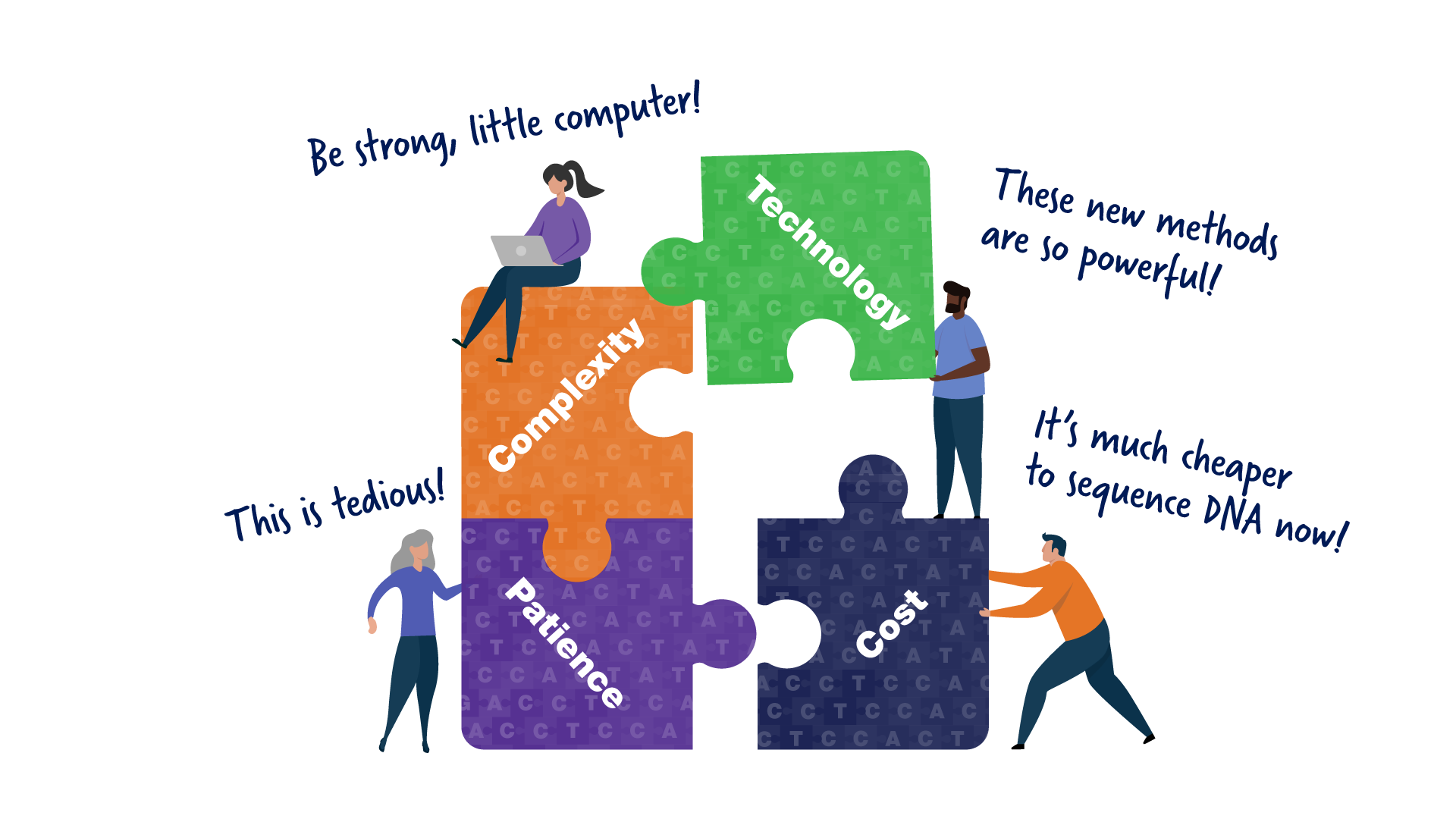 Même avec les nouvelles technologies, le séquençage du génome reste un travail difficile et chronophage qui nécessite beaucoup de compétences et de dévouement. La génération actuelle de chercheurs en génomique est de vrais perfectionnistes et a tout réuni pour enfin compléter la séquence du génome humain.
Même avec les nouvelles technologies, le séquençage du génome reste un travail difficile et chronophage qui nécessite beaucoup de compétences et de dévouement. La génération actuelle de chercheurs en génomique est de vrais perfectionnistes et a tout réuni pour enfin compléter la séquence du génome humain.
https://www.scientificamerican.com/article/completing-the-human-genome-sequence-again/
https://www.genome.gov/about-genomics/telomere-to-telomere
https://news.ucsc.edu/2022/03/t2t-genome.html