 La séquence complète d’un génome humain
La séquence complète d’un génome humain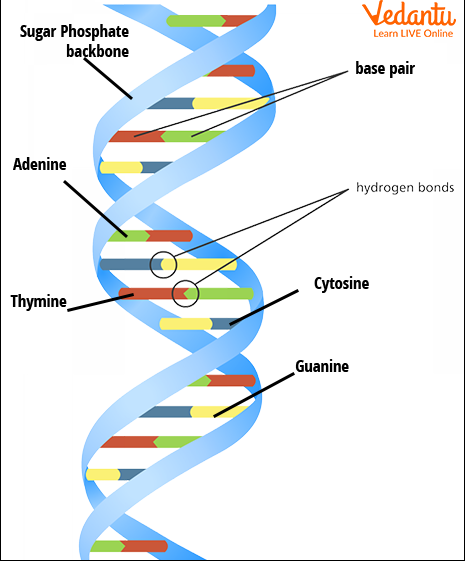 Génomes humains, publics et privés
Génomes humains, publics et privés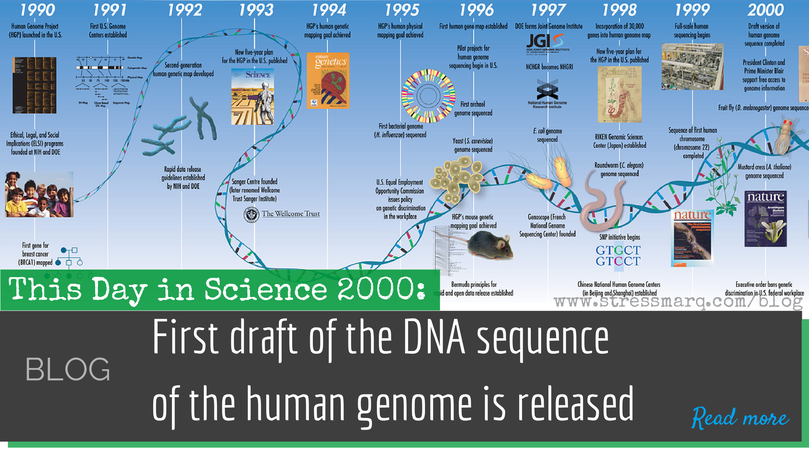 Le secteur commercial en plein essor basé sur l’information génomique pose un défi aux normes de publication scientifique. Mais il reste à établir que les conditions d’accès aux données de séquences publiées doivent changer.
Le secteur commercial en plein essor basé sur l’information génomique pose un défi aux normes de publication scientifique. Mais il reste à établir que les conditions d’accès aux données de séquences publiées doivent changer.![]() La séquence du génome humain contient le code génétique qui se trouve au cœur de chacune des dix billions de cellules de chaque être humain. Elle influence profondément notre corps, notre comportement et notre esprit ; il aidera à l’étude des influences non génétiques sur le développement humain ; cela ouvrira de nouvelles perspectives sur nos origines et notre histoire en tant qu’espèce ; et il indique de nouvelles façons de combattre la maladie. Les habitants de nombreux pays ont investi dans la détermination de la séquence du projet du génome humain, et il est difficile de voir comment cet investissement aurait pu obtenir de meilleurs rendements. Après avoir publié ses données quotidiennement dès le départ avec un accès illimité, le consortium financé par des fonds publics a assemblé environ 92 % de la séquence.
La séquence du génome humain contient le code génétique qui se trouve au cœur de chacune des dix billions de cellules de chaque être humain. Elle influence profondément notre corps, notre comportement et notre esprit ; il aidera à l’étude des influences non génétiques sur le développement humain ; cela ouvrira de nouvelles perspectives sur nos origines et notre histoire en tant qu’espèce ; et il indique de nouvelles façons de combattre la maladie. Les habitants de nombreux pays ont investi dans la détermination de la séquence du projet du génome humain, et il est difficile de voir comment cet investissement aurait pu obtenir de meilleurs rendements. Après avoir publié ses données quotidiennement dès le départ avec un accès illimité, le consortium financé par des fonds publics a assemblé environ 92 % de la séquence.
La séquence complète d’un génome humain Depuis sa publication initiale en 2000, le génome humain de référence n’a couvert que la fraction euchromatique du génome, laissant d’importantes régions hétérochromatiques inachevées. S’adressant aux 8 % restants du génome, le Consortium Telomere-to-Telomer (T2T) présente une séquence complète de 3,055 milliards de paires de bases d’un génome humain, T2T-CHM13, qui comprend des assemblages sans interruption pour tous les chromosomes sauf Y, corrige les erreurs dans les références antérieures, et introduit près de 200 millions de paires de bases de séquence contenant 1956 prédictions de gènes, dont 99 sont prédites comme codant pour des protéines. Les régions complétées comprennent tous les réseaux satellites centromériques, les duplications segmentaires récentes et les bras courts des cinq chromosomes acrocentriques, ouvrant ces régions complexes du génome à des études variationnelles et fonctionnelles.
Depuis sa publication initiale en 2000, le génome humain de référence n’a couvert que la fraction euchromatique du génome, laissant d’importantes régions hétérochromatiques inachevées. S’adressant aux 8 % restants du génome, le Consortium Telomere-to-Telomer (T2T) présente une séquence complète de 3,055 milliards de paires de bases d’un génome humain, T2T-CHM13, qui comprend des assemblages sans interruption pour tous les chromosomes sauf Y, corrige les erreurs dans les références antérieures, et introduit près de 200 millions de paires de bases de séquence contenant 1956 prédictions de gènes, dont 99 sont prédites comme codant pour des protéines. Les régions complétées comprennent tous les réseaux satellites centromériques, les duplications segmentaires récentes et les bras courts des cinq chromosomes acrocentriques, ouvrant ces régions complexes du génome à des études variationnelles et fonctionnelles.
Le génome de référence humain actuel a été publié par le Consortium de référence du génome (GRC) en 2013 et plus récemment corrigé en 2019. Cette référence trouve son origine dans le projet du génome humain financé par des fonds publics et a été continuellement améliorée au cours des deux dernières décennies. Contrairement à l’effort concurrent de Celera et à la plupart des projets de séquençage modernes basés sur l’assemblage de séquences «fusil de chasse», l’assemblage GRC a été construit à partir de chromosomes artificiels bactériens (BAC) séquencés qui ont été ordonnés et orientés le long du génome humain au moyen d’un hybride de rayonnement, d’une liaison génétique et cartes d’empreintes digitales. Cependant, les limites du clonage BAC ont conduit à une sous-représentation des séquences répétitives, et l’assemblage opportuniste de BAC dérivés de plusieurs individus a abouti à une mosaïque d’haplotypes. En conséquence, plusieurs lacunes d’assemblage GRC sont insolubles en raison de polymorphismes structurels incompatibles sur leurs flancs, et de nombreuses autres régions répétitives et polymorphes ont été laissées inachevées ou mal assemblées.
L’assemblage de référence GRCh38 contient 151 méga-paires de bases (Mbp) de séquence inconnue réparties dans tout le génome, y compris les régions péricentromériques et subtélomériques, les duplications segmentaires récentes, les réseaux de gènes ampliconiques et les réseaux d’ADN ribosomal (ADNr), qui sont tous nécessaires pour les fondamentaux. Processus cellulaires.  Certaines des plus grandes lacunes de référence comprennent les réseaux de répétition de satellites humains (HSat) et les bras courts des cinq chromosomes acrocentriques, qui sont représentés dans GRCh38 sous forme d’étendues multimégabases de bases inconnues. En plus de ces lacunes apparentes, d’autres régions de GRCh38 sont artificielles ou sont autrement incorrectes. Par exemple, les réseaux de satellites alpha centromériques sont représentés comme des modèles générés par calcul de monomères satellites alpha pour servir de leurres pour les analyses de reséquençage, et la séquence attribuée au bras court du chromosome 21 apparaît faussement dupliquée et mal assemblée. Comparé à d’autres génomes humains, GRCh38 montre également un biais de suppression à l’échelle du génome qui indique un assemblage incomplet. Malgré les efforts de finition du projet du génome humain et du GRC qui ont amélioré la qualité de la référence, il y a eu des progrès limités pour combler les lacunes restantes dans les années qui ont suivi.
Certaines des plus grandes lacunes de référence comprennent les réseaux de répétition de satellites humains (HSat) et les bras courts des cinq chromosomes acrocentriques, qui sont représentés dans GRCh38 sous forme d’étendues multimégabases de bases inconnues. En plus de ces lacunes apparentes, d’autres régions de GRCh38 sont artificielles ou sont autrement incorrectes. Par exemple, les réseaux de satellites alpha centromériques sont représentés comme des modèles générés par calcul de monomères satellites alpha pour servir de leurres pour les analyses de reséquençage, et la séquence attribuée au bras court du chromosome 21 apparaît faussement dupliquée et mal assemblée. Comparé à d’autres génomes humains, GRCh38 montre également un biais de suppression à l’échelle du génome qui indique un assemblage incomplet. Malgré les efforts de finition du projet du génome humain et du GRC qui ont amélioré la qualité de la référence, il y a eu des progrès limités pour combler les lacunes restantes dans les années qui ont suivi. Le séquençage de fusil de chasse à lecture longue surmonte les limites de l’assemblage basé sur BAC et contourne les défis du polymorphisme structurel entre les génomes. Les lectures multikilobases et monomoléculaires de PacBio se sont avérées capables de résoudre des variations structurelles complexes et des lacunes dans GRCh38, tandis que les lectures « ultralongues » > 100 kbp d’Oxford Nanopore ont permis des assemblages complets d’un centromère humain (chromosome Y) et, plus tard, d’un chromosome entier (chromosome X). Cependant, le taux d’erreur élevé (> 5%) de ces technologies a posé des défis pour l’assemblage de réseaux de répétition longs et presque identiques. Le plus récent séquençage de consensus circulaire «HiFi» de PacBio offre un compromis de longueurs de lecture de 20 kbp avec un taux d’erreur de 0,1 %. Alors que les lectures ultra longues sont utiles pour couvrir les répétitions, les lectures HiFi excellent pour différencier les copies répétées ou les haplotypes subtilement divergents.
Le séquençage de fusil de chasse à lecture longue surmonte les limites de l’assemblage basé sur BAC et contourne les défis du polymorphisme structurel entre les génomes. Les lectures multikilobases et monomoléculaires de PacBio se sont avérées capables de résoudre des variations structurelles complexes et des lacunes dans GRCh38, tandis que les lectures « ultralongues » > 100 kbp d’Oxford Nanopore ont permis des assemblages complets d’un centromère humain (chromosome Y) et, plus tard, d’un chromosome entier (chromosome X). Cependant, le taux d’erreur élevé (> 5%) de ces technologies a posé des défis pour l’assemblage de réseaux de répétition longs et presque identiques. Le plus récent séquençage de consensus circulaire «HiFi» de PacBio offre un compromis de longueurs de lecture de 20 kbp avec un taux d’erreur de 0,1 %. Alors que les lectures ultra longues sont utiles pour couvrir les répétitions, les lectures HiFi excellent pour différencier les copies répétées ou les haplotypes subtilement divergents.
Pour terminer les dernières régions restantes du génome, nous avons tiré parti des aspects complémentaires du séquençage à lecture ultralongue PacBio HiFi et Oxford Nanopore pour assembler la lignée cellulaire CHM13hTERT uniformément homozygote (ci-après, CHM13). L’assemblage de référence T2T-CHM13 qui en résulte supprime une barrière vieille de 20 ans qui a caché 8% du génome de l’analyse basée sur la séquence, y compris toutes les régions centromériques et les bras courts entiers de cinq chromosomes humains. Ici, nous décrivons la construction, la validation et l’analyse initiale d’un génome de référence humain vraiment complet et discutons de son impact potentiel sur le terrain. Lignée cellulaire et séquençage
Lignée cellulaire et séquençage Comme pour de nombreux efforts d’amélioration du génome de référence antérieurs, y compris les assemblages T2T des chromosomes humains X et 8, nous avons ciblé une taupe hydatiforme complète (CHM) pour le séquençage. La plupart des génomes CHM proviennent de la perte du complément maternel et de la duplication du complément paternel après la fécondation et sont donc homozygotes avec un caryotype 46, XX. Le séquençage de CHM13 a confirmé une homozygotie presque uniforme, à l’exception de quelques milliers de variants hétérozygotes et d’une délétion hétérozygote à l’échelle de la mégabase dans le réseau d’ADNr sur le chromosome 15. L’analyse de l’ascendance locale montre que la majeure partie du génome CHM13 est d’origine européenne, y compris les régions d’introgression néandertalienne, avec un certain mélange prédit (Fig. 1A). Par rapport à divers échantillons du projet 1000 Genomes (1KGP), CHM13 ne possède aucun excès apparent d’allèles singletons ou de variantes de perte de fonction.
Comme pour de nombreux efforts d’amélioration du génome de référence antérieurs, y compris les assemblages T2T des chromosomes humains X et 8, nous avons ciblé une taupe hydatiforme complète (CHM) pour le séquençage. La plupart des génomes CHM proviennent de la perte du complément maternel et de la duplication du complément paternel après la fécondation et sont donc homozygotes avec un caryotype 46, XX. Le séquençage de CHM13 a confirmé une homozygotie presque uniforme, à l’exception de quelques milliers de variants hétérozygotes et d’une délétion hétérozygote à l’échelle de la mégabase dans le réseau d’ADNr sur le chromosome 15. L’analyse de l’ascendance locale montre que la majeure partie du génome CHM13 est d’origine européenne, y compris les régions d’introgression néandertalienne, avec un certain mélange prédit (Fig. 1A). Par rapport à divers échantillons du projet 1000 Genomes (1KGP), CHM13 ne possède aucun excès apparent d’allèles singletons ou de variantes de perte de fonction.
Nous avons séquencé de manière approfondie CHM13 avec plusieurs technologies, y compris le séquençage consensuel circulaire PacBio 30 × (HiFi), le séquençage à lecture ultralongue (ONT) 120 × Oxford Nanopore, 100 × Illumina PCR-Free séquençage (ILMN), 70 × Illumina Arima Genomics Hi-C (Hi-C), cartes optiques BioNano et séquençage de brin de matrice d’ADN unicellulaire (Strand-seq) (tableau S1). Pour permettre l’assemblage des réseaux de satellites centromériques hautement répétitifs et des duplications segmentaires étroitement liées, nous avons développé des méthodes d’assemblage, de polissage et de validation qui utilisent mieux ces ensembles de données disponibles. Assemblage du génome La base de l’assemblage T2T-CHM13 est un graphe de chaînes d’assemblage haute résolution construit directement à partir de lectures HiFi. Dans un graphe de chaînes bidirectionnel, les nœuds représentent des séquences assemblées sans ambiguïté et les arêtes correspondent aux chevauchements entre eux, en raison de répétitions ou de véritables adjacences dans le génome sous-jacent. Le graphe CHM13 a été construit à l’aide d’une méthode spécialement conçue qui combine des composants d’assembleurs existants avec un traitement de graphe spécialisé.
Assemblage du génome La base de l’assemblage T2T-CHM13 est un graphe de chaînes d’assemblage haute résolution construit directement à partir de lectures HiFi. Dans un graphe de chaînes bidirectionnel, les nœuds représentent des séquences assemblées sans ambiguïté et les arêtes correspondent aux chevauchements entre eux, en raison de répétitions ou de véritables adjacences dans le génome sous-jacent. Le graphe CHM13 a été construit à l’aide d’une méthode spécialement conçue qui combine des composants d’assembleurs existants avec un traitement de graphe spécialisé. 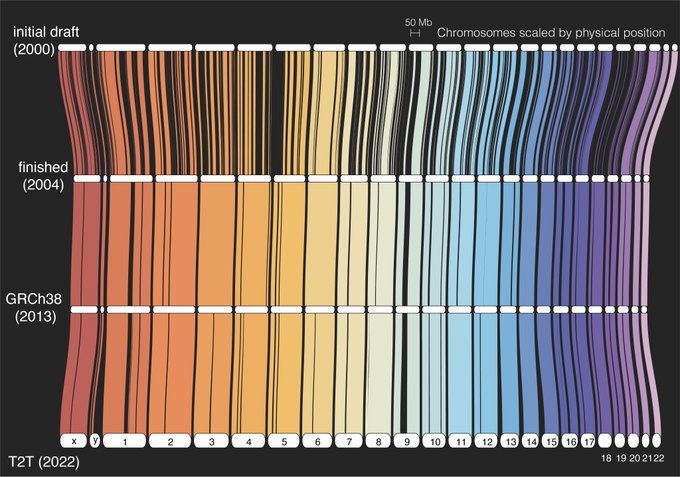
Le Consortium international de séquençage du génome humain décrit la séquence finale du génome humain 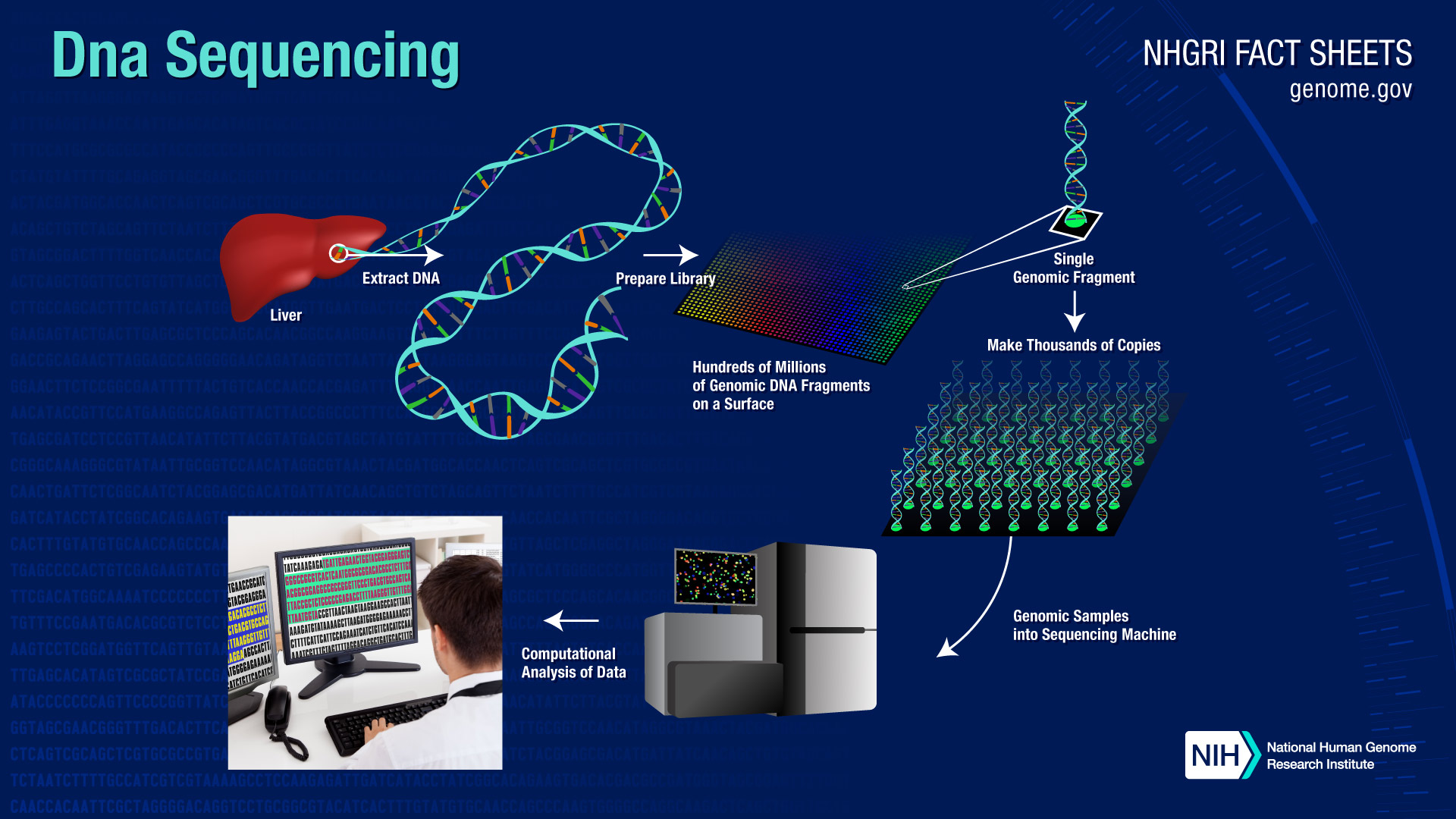
Des chercheurs réduisent le nombre de gènes humains à 20 000-25 000![]() BETHESDA, Maryland, mercredi 20 octobre 2004 – Le Consortium international de séquençage du génome humain, dirigé aux États-Unis par l’Institut national de recherche sur le génome humain (NHGRI) et le Département de l’énergie (DOE), a publié aujourd’hui sa description scientifique de la séquence finale du génome humain, réduisant le nombre estimé de gènes codant pour les protéines humaines de 35 000 à seulement 20 000-25 000, un nombre étonnamment bas pour notre espèce. L’article paraît dans le numéro du 21 octobre de la revue Nature. Dans l’article, les chercheurs décrivent le produit final du projet du génome humain, qui était l’effort de 13 ans pour lire les informations codées dans les chromosomes humains qui a atteint son point culminant en 2003. La publication fournit des preuves scientifiques rigoureuses que la séquence du génome produite par le projet du génome humain a à la fois la couverture élevée et la précision nécessaires pour effectuer des analyses sensibles, telles que la concentration sur le nombre de gènes, les duplications segmentaires impliquées dans la maladie et la « naissance » et » mort » des gènes au cours de l’évolution.
BETHESDA, Maryland, mercredi 20 octobre 2004 – Le Consortium international de séquençage du génome humain, dirigé aux États-Unis par l’Institut national de recherche sur le génome humain (NHGRI) et le Département de l’énergie (DOE), a publié aujourd’hui sa description scientifique de la séquence finale du génome humain, réduisant le nombre estimé de gènes codant pour les protéines humaines de 35 000 à seulement 20 000-25 000, un nombre étonnamment bas pour notre espèce. L’article paraît dans le numéro du 21 octobre de la revue Nature. Dans l’article, les chercheurs décrivent le produit final du projet du génome humain, qui était l’effort de 13 ans pour lire les informations codées dans les chromosomes humains qui a atteint son point culminant en 2003. La publication fournit des preuves scientifiques rigoureuses que la séquence du génome produite par le projet du génome humain a à la fois la couverture élevée et la précision nécessaires pour effectuer des analyses sensibles, telles que la concentration sur le nombre de gènes, les duplications segmentaires impliquées dans la maladie et la « naissance » et » mort » des gènes au cours de l’évolution.
« Il y a seulement une décennie, la plupart des scientifiques pensaient que les humains avaient environ 100 000 gènes. Lorsque nous avons analysé l’ébauche de travail de la séquence du génome humain il y a trois ans, nous avons estimé qu’il y avait environ 30 000 à 35 000 gènes, ce qui en a surpris beaucoup. Cette nouvelle analyse réduit ce nombre encore plus loin et nous fournit l’image la plus claire à ce jour de notre génome », a déclaré le directeur du NHGRI, Francis S. Collins, MD, PhD. « La disponibilité de la séquence très précise du génome humain dans des bases de données publiques gratuites permet aux chercheurs du monde entier de mener des études encore plus précises de notre livre d’instructions génétiques et de son influence sur la santé et la maladie. » L’un des objectifs centraux de l’effort d’analyse du génome humain est l’identification de tous les gènes, qui sont généralement définis comme des segments d’ADN qui codent pour des protéines particulières. Selon les nouvelles découvertes, les chercheurs ont confirmé l’existence de 19 599 gènes codant pour des protéines dans le génome humain et identifié 2 188 autres segments d’ADN qui devraient être des gènes codant pour des protéines.
« L’analyse a révélé que certains des modèles de gènes antérieurs étaient erronés en raison de défauts dans la séquence préliminaire inachevée du génome humain », a déclaré Jane Rogers, PhD., responsable du séquençage au Wellcome Trust Sanger Institute à Hinxton, en Angleterre. . « La tâche d’identification des gènes reste difficile, mais a été grandement facilitée par la séquence du génome humain finie, ainsi que par la disponibilité de séquences du génome d’autres organismes, de meilleurs modèles informatiques et d’autres ressources améliorées. » L’article de Nature fournit également à la communauté scientifique une description révisée par des pairs du processus de finition et une évaluation de la qualité de la séquence du génome humain finie, qui a été déposée dans des bases de données publiques en avril 2003. L’évaluation confirme que la séquence finie est maintenant couvre plus de 99 % de la partie euchromatique (ou contenant des gènes) du génome humain et a été séquencé avec une précision de 99,999 %, ce qui se traduit par un taux d’erreur de seulement 1 base pour 100 000 paires de bases – 10 fois plus précis que le objectif originel.
La contiguïté de la séquence est aussi massivement améliorée. La lettre d’ADN moyenne repose désormais sur une étendue de 38,5 millions de paires de bases de séquence ininterrompue de haute qualité – environ 475 fois plus longue que l’étendue de 81 500 paires de bases qui était disponible dans le projet de travail.
« La séquence du génome humain dépasse de loin nos attentes en termes de précision, d’exhaustivité et de continuité. Elle reflète le dévouement de centaines de scientifiques travaillant ensemble vers un objectif commun : créer une base solide pour la biomédecine au 21e siècle », a déclaré Eric Lander, PhD., directeur du Broad Institute du MIT et de Harvard à Cambridge, Mass. En plus de réduire le nombre de gènes humains, les scientifiques ont rapporté que l’amélioration de la qualité de la séquence du génome humain finie, par rapport aux ébauches précédentes, fournit une image beaucoup plus claire de certains phénomènes tels que la duplication de segments d’ADN et la naissance et la mort de gènes.
Les duplications segmentaires sont de grandes copies presque identiques d’ADN, qui sont présentes à au moins deux endroits du génome humain. Un certain nombre de maladies humaines sont connues pour être associées à des mutations dans des régions dupliquées par segments, notamment le syndrome de Williams, le syndrome de Charcot-Marie-Tooth et le syndrome de DiGeorge. « Les duplications segmentaires étaient presque impossibles à étudier dans le projet de séquence. Maintenant, grâce aux efforts inlassables de groupes du monde entier, cette partie importante et en évolution rapide de notre génome est ouverte à l’exploration scientifique », a déclaré Robert H. Waterston, MD, PhD., ancien directeur du Centre de séquençage du génome de l’Université de Washington à St. Louis et maintenant directeur du Département des sciences du génome de l’Université de Washington à Seattle.
« Les duplications segmentaires étaient presque impossibles à étudier dans le projet de séquence. Maintenant, grâce aux efforts inlassables de groupes du monde entier, cette partie importante et en évolution rapide de notre génome est ouverte à l’exploration scientifique », a déclaré Robert H. Waterston, MD, PhD., ancien directeur du Centre de séquençage du génome de l’Université de Washington à St. Louis et maintenant directeur du Département des sciences du génome de l’Université de Washington à Seattle.
Les duplications segmentaires couvrent 5,3 % du génome humain, bien plus que dans le génome du rat, qui en compte environ 3 %, ou que le génome de la souris, qui en compte entre 1 et 2 %. Les duplications segmentaires permettent de comprendre comment notre génome a évolué et continue de changer. La forte proportion de duplication segmentaire dans le génome humain montre que notre matériel génétique a subi une innovation fonctionnelle et un changement structurel rapides au cours des 40 derniers millions d’années, contribuant vraisemblablement à des caractéristiques uniques qui nous séparent de nos ancêtres primates non humains.
L’analyse du consortium a révélé que la distribution des duplications segmentaires varie considérablement d’un chromosome humain à l’autre. Le chromosome Y est le cas le plus extrême, avec des duplications segmentaires sur plus de 25 % de sa longueur. Certaines duplications segmentaires ont tendance à être regroupées près du milieu (centromères) et des extrémités (télomères) de chaque chromosome, où, selon les chercheurs, elles peuvent être utilisées par le génome comme laboratoire évolutif pour créer des gènes dotés de nouvelles fonctions.La précision de la séquence finale du génome humain produite par le projet du génome humain a également donné aux scientifiques un aperçu initial de la naissance et de la mort des gènes dans le génome humain. Les scientifiques ont identifié plus de 1 000 nouveaux gènes apparus dans le génome humain après notre divergence avec les rongeurs il y a environ 75 millions d’années. La plupart d’entre eux sont issus de duplications génétiques récentes et sont impliqués dans les fonctions immunitaires, olfactives et reproductives. Par exemple, il existe deux familles de gènes récemment dupliqués dans le génome humain qui codent pour des ensembles de protéines (glycoprotéine bêta-1 spécifique à la grossesse et protéines bêta de la choriogonadotropine) qui peuvent être impliquées dans la période prolongée de grossesse propre aux humains.
De plus, les chercheurs ont utilisé le génome humain fini pour identifier et caractériser 33 gènes presque intacts qui ont récemment acquis une ou plusieurs mutations, les obligeant à cesser de fonctionner ou à « mourir ». Les scientifiques ont identifié ces gènes non fonctionnels, appelés pseudogènes, dans le génome humain en les alignant avec les génomes de la souris et du rat, dans lesquels les gènes correspondants ont conservé leur fonctionnalité. Fait intéressant, les chercheurs ont déterminé que 10 de ces pseudogènes dans la séquence du génome humain semblent avoir codé des protéines impliquées dans la réception olfactive, ce qui aide à expliquer pourquoi les humains ont moins de récepteurs olfactifs fonctionnels et, par conséquent, un odorat plus faible que les rongeurs.
Ensuite, les chercheurs ont aligné les 33 pseudogènes avec le brouillon de séquence du génome du chimpanzé pour déterminer s’ils étaient encore fonctionnels avant la divergence d’Homo sapiens avec les grands singes il y a environ 5 millions d’années. L’analyse a révélé que 27 des pseudogènes n’étaient pas fonctionnels chez les humains et les chimpanzés. Cependant, cinq des gènes qui étaient inactifs chez l’homme se sont avérés encore fonctionnels chez les chimpanzés.  « L’identification de ces pseudogènes et de leurs homologues fonctionnels chez le chimpanzé fournit un terrain fertile pour de futurs projets de recherche », a déclaré Richard Gibbs, PhD., directeur du centre de séquençage du génome humain du Baylor College of Medicine à Houston, qui séquence actuellement le génome de un autre primate non humain, le macaque rhésus (Macaca mulatta). Plus de 2 800 chercheurs qui ont participé au Consortium international de séquençage du génome humain partagent la paternité de l’article Nature d’aujourd’hui, qui développe l’analyse initiale du groupe publiée en février 2001. Des annotations et des analyses encore plus détaillées ont déjà été publiées pour les chromosomes 5, 6, 7, 9, 10, 13, 14, 19, 20, 21, 22 et Y. Des publications décrivant les 12 chromosomes restants sont à venir.
« L’identification de ces pseudogènes et de leurs homologues fonctionnels chez le chimpanzé fournit un terrain fertile pour de futurs projets de recherche », a déclaré Richard Gibbs, PhD., directeur du centre de séquençage du génome humain du Baylor College of Medicine à Houston, qui séquence actuellement le génome de un autre primate non humain, le macaque rhésus (Macaca mulatta). Plus de 2 800 chercheurs qui ont participé au Consortium international de séquençage du génome humain partagent la paternité de l’article Nature d’aujourd’hui, qui développe l’analyse initiale du groupe publiée en février 2001. Des annotations et des analyses encore plus détaillées ont déjà été publiées pour les chromosomes 5, 6, 7, 9, 10, 13, 14, 19, 20, 21, 22 et Y. Des publications décrivant les 12 chromosomes restants sont à venir.
La séquence du génome humain finie et ses annotations sont accessibles via les navigateurs de génome publics suivants. :GenBank ( www.ncbi.nih.gov/Genbank ) au National Center for Biotechnology Information (NCBI) des NIH ; le UCSC Genome Browser ( www.genome.ucsc.edu ) à l’Université de Californie à Santa Cruz ; l’Ensembl Genome Browser ( www.ensembl.org ) au Wellcome Trust Sanger Institute et à l’EMBL-European Bioinformatics Institute ; la Banque de données ADN du Japon ( www.ddbj.nih.ac.jp ); et EMBL-Bank ( www.ebi.ac.uk/embl/index.html ) dans la base de données de séquences de nucléotides du Laboratoire européen de biologie moléculaire.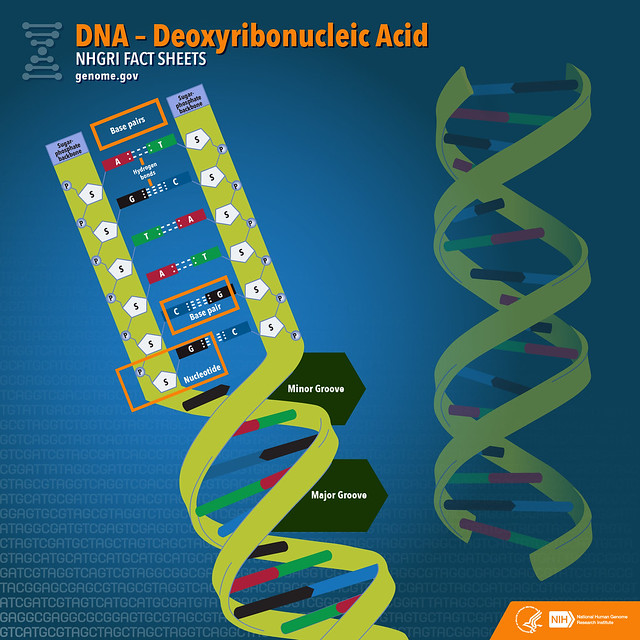 Le Consortium international de séquençage du génome humain regroupe des scientifiques de 20 institutions situées en France, en Allemagne, au Japon, en Chine, en Grande-Bretagne et aux États-Unis. Les cinq plus grands centres de séquençage sont situés au : Baylor College of Medicine ; le Broad Institute du MIT et de Harvard ; Joint Genome Institute du DOE, Walnut Creek, Californie ; École de médecine de l’Université de Washington ; et le Wellcome Trust Sanger Institute.
Le Consortium international de séquençage du génome humain regroupe des scientifiques de 20 institutions situées en France, en Allemagne, au Japon, en Chine, en Grande-Bretagne et aux États-Unis. Les cinq plus grands centres de séquençage sont situés au : Baylor College of Medicine ; le Broad Institute du MIT et de Harvard ; Joint Genome Institute du DOE, Walnut Creek, Californie ; École de médecine de l’Université de Washington ; et le Wellcome Trust Sanger Institute.
Le projet 1000 génomes – D’un génome à 1000 et au-delà en 25 ans  Nous célébrons le 25 e anniversaire du lancement du Human Genome Project (HGP) avec la reconnaissance des ressources en génomique humaine, d’un génome à plus de mille génomes et au-delà. Le projet 1000 Genomes a débuté en 2007 dans le but de développer une ressource complète de la variation génétique humaine à travers les populations mondiales. Huit ans plus tard, nous publions dans ce numéro les rapports de la phase finale de ce projet, représentant l’évaluation la plus complète de la variation génétique humaine dans les populations mondiales à ce jour.
Nous célébrons le 25 e anniversaire du lancement du Human Genome Project (HGP) avec la reconnaissance des ressources en génomique humaine, d’un génome à plus de mille génomes et au-delà. Le projet 1000 Genomes a débuté en 2007 dans le but de développer une ressource complète de la variation génétique humaine à travers les populations mondiales. Huit ans plus tard, nous publions dans ce numéro les rapports de la phase finale de ce projet, représentant l’évaluation la plus complète de la variation génétique humaine dans les populations mondiales à ce jour.  Les ensembles de données déjà établis ont, depuis leur lancement, fourni une ressource ouverte fondamentale qui a permis une multitude d’associations génétiques robustes à la maladie ainsi que de nombreuses informations clés sur l’histoire et l’évolution de la population.
Les ensembles de données déjà établis ont, depuis leur lancement, fourni une ressource ouverte fondamentale qui a permis une multitude d’associations génétiques robustes à la maladie ainsi que de nombreuses informations clés sur l’histoire et l’évolution de la population. 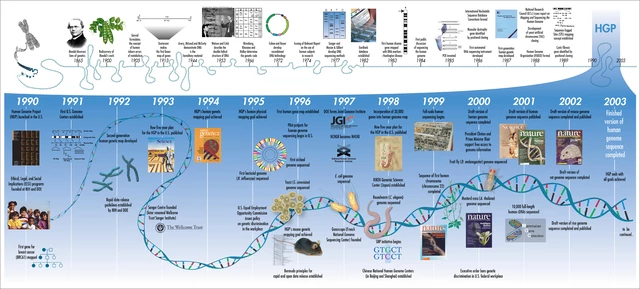 Les publications de la phase finale du projet 1000 Genomes représentent non seulement l’achèvement de ce projet, mais aussi l’aboutissement d’une série de collaborations internationales issues du HGP, y compris le projet international HapMap, toutes axées sur l’établissement de catalogues de référence ouverts de la variation génétique en tant que ressource. à la communauté. Nous sommes heureux de présenter cette collection Nature de toutes les publications principales et des nouvelles et commentaires connexes sur les projets internationaux HapMap et 1000 Génomes.
Les publications de la phase finale du projet 1000 Genomes représentent non seulement l’achèvement de ce projet, mais aussi l’aboutissement d’une série de collaborations internationales issues du HGP, y compris le projet international HapMap, toutes axées sur l’établissement de catalogues de référence ouverts de la variation génétique en tant que ressource. à la communauté. Nous sommes heureux de présenter cette collection Nature de toutes les publications principales et des nouvelles et commentaires connexes sur les projets internationaux HapMap et 1000 Génomes.
https://www.genome.gov/12513430/2004-release-ihgsc-describes-finished-human-sequence